دانلود پروژه ورد ساختار ميتوكندري در انسان و نقش آن در ايجاد بيماريهاي مختلف ميتوكندري
مقدمه:
اولين گزارشات در ارتباط با ساختارهاي درون سلولي شبه ميتوكندري به ۱۵۰ سال پيش برميگردد. واژه ميتوكندري كه از دو كلمه يوناني mitos بمعني نخ يا رشته و chondros به معني گرانول منشا گرفته است؛ براي اولين بار صد سال پيش مورد استفاده قرار گرفت. عملكرد اصلي اين ارگانل كروي يا ميلهاي شكل كه صدها عدد از آن در يك سلول وجود دارد، فسفريلاسيون اكسيداتيو است؛ بعبارت ديگر اكسيداسيون سوبستراها به Co2 و آب و فراهم كردن تركيب پرانرژي ATP براي سلولها؛ و به همين دليل است كه ميتوكندري را نيروگاه يا موتورخانه سلول نيز مينامند. بيماريهاي دژنراتيو بسيار زيادي تا به امروز با نارساييها و اختلالات ميتوكندري مرتبط شدهاند. اين بيماريها ميتوانند در اثر موتاسيون در DNA ميتوكندري و يا DNA هسته ايجاد شوند. اولين بيماريهاي ميتوكندريايي كه در سطح ملكولي درك شدند؛ در يك بيمار CPEO (فلج مزمن پيشرونده عضلات چشمي خارجي) و KSS (سندرمkearns-sayre) گزارش شدند. در همان زمان wallace موتاسيوني نقطهاي را در ژن ND6 گزارش كرد كه با LHON (نوروپاتي چشمي ارثي لبر) مرتبط است. در سال ۱۹۹۰، دوموتاسيون جديد، يكي در ژن لايزيل- tRNA در سندرم MERRF و ديگري در ژن لوسيل – tRNA در سندرم MELAS گزارش شدند. طيف فتوتيپي بيماريهاي ميتوكندريايي از ميوپاتيهاي نادر تا بيماريهاي متعدد را شامل ميشود. برخي موتاسيونهاي mtDNA، علائم و نشانههاي منحصر و ويژهاي دارند؛ مثل جهشهاي اشتباهي كه موجب نوروپاتي چشمي ارثي لبر ميشوند در حاليكه بقيه تظاهرات مولتي سيستم متنوعي را شامل ميشوند مثل جهشهاي حذفي كه موجب CPEO ميشوند. بيماريهاي ميتوكندريايي بواسطه وراثت مادري، وراثت منرلي و نيز نوتركيبيهاي دوتايي نو، قادر به انتقال ميباشند. اين پيچيدگي ژنتيكي از اين حقيقت ناشي ميشود كه ميتوكندري از حدود ۱۰۰۰ ژن كه در بين ژنوم ميتوكندري و هسته پخش شدهاند، تشكيل شده است. علاوه بر اين بيماريهاي ميتوكندريايي غالباً شروع تاخيري و يك دوره پيش رونده دارند كه احتمالاً از تجمع جهشهاي سوماتيك mtDNA در بافتهاي post-mitotic حاصل شدهاند. اين موتاسيونهاي سوماتيك mtDNA همچنين در سرطان و پيري نيز نقش دارند. اگرچه بيماريهاي ميتوكندريايي هر ارگاني را ممكن است درگير كنند اما اين بيماريها غالباً CNS، عضلات اسكلتي، قلب، كليه و سيستمهاي اندوكرين را تحت تاثير قرار ميدهند. علت اين پيچيدگيهاي فتوتيپي، نقش مهم ميتوكندري در انواع پروسههاي سلولي شامل توليد انرژي سلولي بوسيله فسفريلاسيون اكيداتيو، توليد گونههاي سمي فعال اكسيژن (ROS) بعنوان يك محصول جانبي در فسفريلاسيون اكسيداتيو و تنظيم شروع آپوپتوزاز طريق فعال شدن نفوذپذيري پورهاي انتقالي ميتوكندري (mtPTP) است. (۱۹، ۲۰ و ۲۴)
ساختار ميتوكندري :
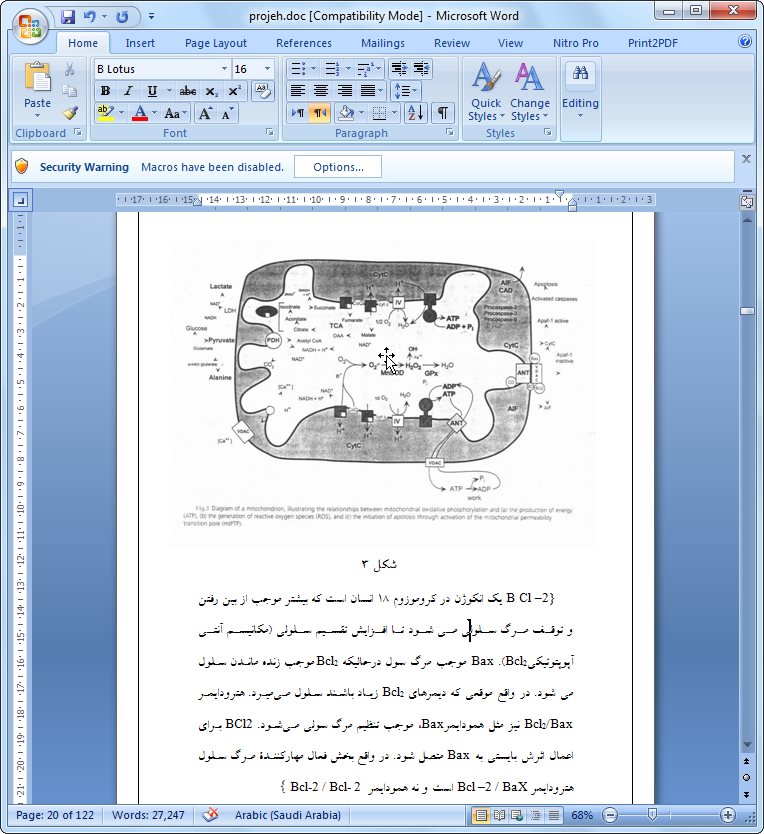
ميتوكندري واجد يك غشاي بيروني و يك غشاي داخلي است كه دو فضاي داخلي را ايجاد ميكنند: ماتريكس داخلي و فضاي بين دو غشا كه بسيار باريك است. غشاي داخلي چينخورده و تعداد زيادي كريستا ايجاد ميكند كه كل سطح آنرا بمقدار زيادي افزايش ميدهد. سطح وسيع غشاي داخلي، آنزيمهاي دستگاه مولد انرژي ميتوكندريايي (زنجيره تنفسي) را در خود جاي داده است. ماتريكس ميتوكندري واجد نسخههاي يكسان متعددي از ژنوم ميتوكندري، ريبوزومهاي ويژه ميتوكندري (ميتوريبوزوم)، tRNAها و آنزيمهاي متنوعي است كه براي بيان ژنهاي ميتوكندري مورد نيازند. (۲۰)
مقدمه
ساختار ميتوكندري
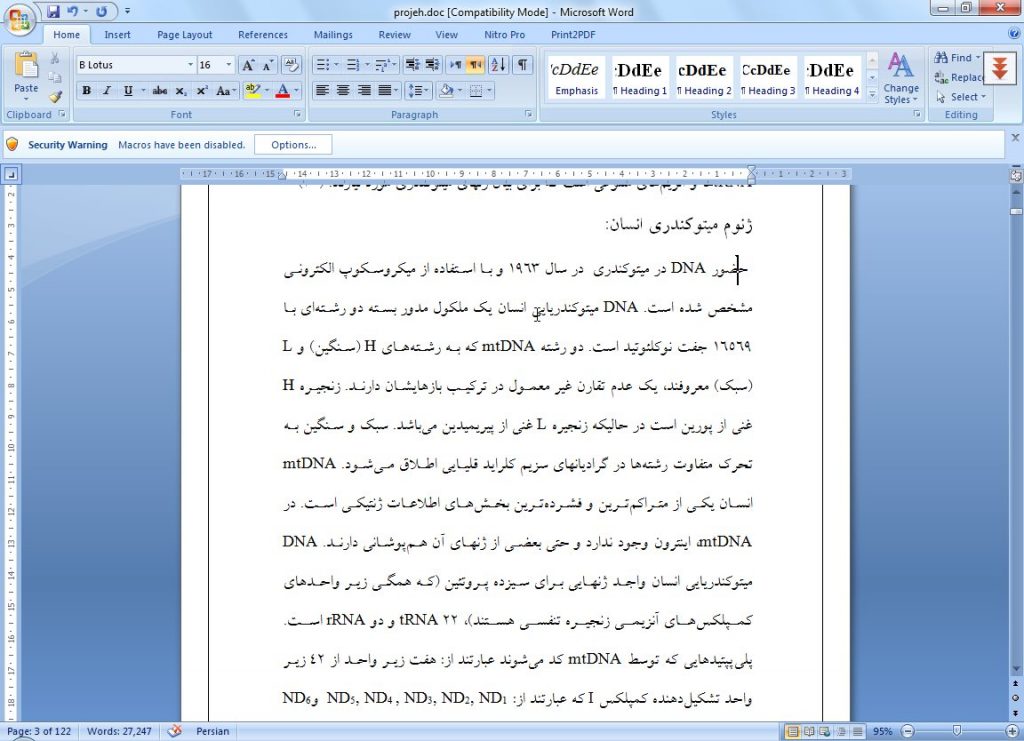
ژنوم ميتوكندري انسان
ميتوكندريها نيمه خود مختار هستند
ميتوكندريها وراثت مادري دارند
هتروپلاسمي و تفكيك رپليكاتيو
نوتركيبي mtDNA
كامل شدن mtDNA
ميزان بالاي موتاسيون در mtDNA
تنوع پلي مورفيك mtDNA در جمعيتهاي انساني
ژنتيك ميتوكندري (همانندسازي، رونويسي و ترجمه mtDNA)
فرايندهاي ميتوكندريايي
ميتوكندري و پاسخ به استرس
بيان آستانهاي
بيماريهاي ميتوكندريايي ناشي از جهشهاي سيستميك
LHON (نوروپاتي چشمي ارثي لبر)
مكانيسمهاي پاتوفيزيولوژيكي احتمالي LHON
LHON، مولتيپل اسكلروزيس و ديستوني
بيماري پاركينسون (PD) و بيماري هانتينگتون (HD)
ژنتيك كروموزومي بيماري پاركينسون
جهشهاي mtDNA در PD
اختلالات ميتوكندريايي در PD
نارساييهاي ميتوكندريايي در بيماري هانتينگتون
رتينيت پيگمنتوزا (RP) و سندرم لي (LS)
موتاسيونهاي mtDNA در RP و سندرم لي
ميوپاتي و انسفالوميوپاتيهاي ميتوكندريايي
ضعف عضلاني پيشرونده و مرتبط پاموتاسيونهاي سيتوكروم mtDNA b
انسفالوميوپاتيهاي ناشي از جهشهاي ژن COX mtDNA
ميوپاتيهاي ميتوكندريايي ناشي از موناسيونهاي TRNA ژنوم ميتوكندري
كارديو ميوپاتيهايپرتروفيك و ميوپاتي ناشي از جهشهاي mtDNA
انسفالوميوپاتيهاي ناشي از جهشهاي mtDNA
افتالموپلژيا، پتوزيس و ميوپاتي ميتوكندريايي
افتالموپلژياي ناشي از جهشهاي mtDNA
CPEO و KSS مرتبط با موتاسيونهاي نوآرايي mtDNA
CPEO ناشي از موتاسيونهاي تعويض باز mtDNA
سندرم مغز استخواني پانكراسي پيرسون
ديابت مليتوس
تيپ II ديابت مليتوس بوسيله نوآراييهاي (حذفها و دوپليكاسيونها) mtDNA
ايجاد ميشود
ديابت تيپ II ناشي از موتاسيونهاي تعويض باز mtDNA
ميوپاتي و ديابت
پاتوفيزيولوژي ديابت و كري
كري به ارث رسيده از مادر و يا كري القا شده توسط آمينوگليكوزيد
دمانس بعنوان يك بيماري ميتوكندريايي
بيولوژي و ژنتيك بيماري آلزايمر
اختلالات ميتوكندريايي در AD
بيماري آلزايمر ناشي از جهشهاي mtDNA
ديس كندروپلاژي متافيزي يا هيپوپلازي مويي- غضروفي ناشي از جهشهاي
RNASE MRP
بيماريهاي مولتي فاكتوريال و mtDNA
جهشهاي سوماتيك mtDNA در بيماريهاي دژنراتيو، سرطان و پيري
تجمع جهشهاي سوماتيك mtDNA مرتبط با سن
آنمي سيدروبلاستيك ايديوپاتيك
بيماري ايسكمي قلبي و كارديوميوپاتي اتساعي
بيماريهاي نورودژنراتيو؛ HD, PD و AD
بيماري پاركينسون و بيماري هانتينگتون
بيماري آلزايمر
موتاسيونهاي سوماتيك mtDNA در ديگر بيماريهاي كمپلكس
موتاسيونهاي سوماتيك در سرطان
نتيجهگيري
منابع
| اطلاعات بیشتر ... | |
|---|---|
| تعداد صفحات | 128 |
| پسوند فایل | doc |
| قابلیت ویرایش | دارد |
| فرمت فایل | ورد |









1 نظر
عالیه به همه دوستان توضیه میکنم حتما بخرید واقعا مفته و خیلی با ارزش